Somatic copy number aberrations (SCNAs) — gains or losses of regions of DNA due to chromosomal instability and structural variations — are prevasive in cancer genomes. High overall ploidy are often observed in the genome of clinical samples which suggests genome doubling. In a tumor with the chromosomal instabilty phenotype, different genomic regions can evolve to distinct integer copy number (CN) states, and tumor subpopulation can differ in the CN states for a particular region. On the other hand, somatic single nucleotide variants (SSNVs, or point mutations) is another crucial means by which cancer genome evolve. As compared to SCNAs, SSNVs are relatively easier to be analyzed and functionally evaluated experimentally. However, it remains a question if these two genomic errors are independent of each other and which of them are more important for the development of the disease? Here, we would like to share our thoughts regarding the impact of SCNAs on the burden of SSNVs.
This is not an a new topic, as we have seen several publications showing the relation between SCNAs and SSNVs. The most famous one (that we know) is the paper in Nature Genetics, where the authors reported that there is a mutual exclusivity between SCNAs and SSNVs, i.e., some tumors tend to be “copy number driven” whereas other are “mutation driven”. In later studies, however, slight positive correlation between aneuploidy score and mutation burden are also reported using large patient cohort. In these papers, the hypermutated tumors — those with exteremly high mutation burdens due to deficiency in mismatch repair mechanisms — are exluded. Whereas knowing the positive correlation is a good thing, but how do we interpret such a pattern?
A research area that jointly analyzes the SSNV and SCNAs is the timing analysis" which aims at reconstructing the evolutionary history of genomic alterations. Pioneering papers in this domain include Greenman’s in Genome Research, Purdom in Bioinformatics, and more recent application of this theorecital concept in large patient cohort published in Nature. In these studies, there was an implicit assumption: the SSNV rate per nucleotide remains constant for a given genomic segment. In other words, a genomic segment resting on a CN state would accumulate SSNVs at a rate proportional to the corresponding number of copies. As a result, site frequency spectrum (SFS) of SSNVs in a DNA segment affected by SCNA depends on the trajectory of the SCNA, i.e., the order and time span on each CN state that the segment has ever rested on. In the same tumor genome, the higher a CN state, and the longer the segment remains on that state, the more SSNVs are expected for the corresponding segment. Such a bias explains the positive correlation between SCNA and SSNVs observed to some extent, but we shoule be caucious in using this to explain the variability between different patients as numours factors can contribute to the rates of SSNV and SCNAs, respectively. Whereas the constant mutation rate in a given segment may not be always valid, we believe that this simple relation seems to be a good null model to start with.
\begin{equation} Mutation burden ~ \sigma(CN at stage k x Time duration for stage k x mutation rate) \end{equation}
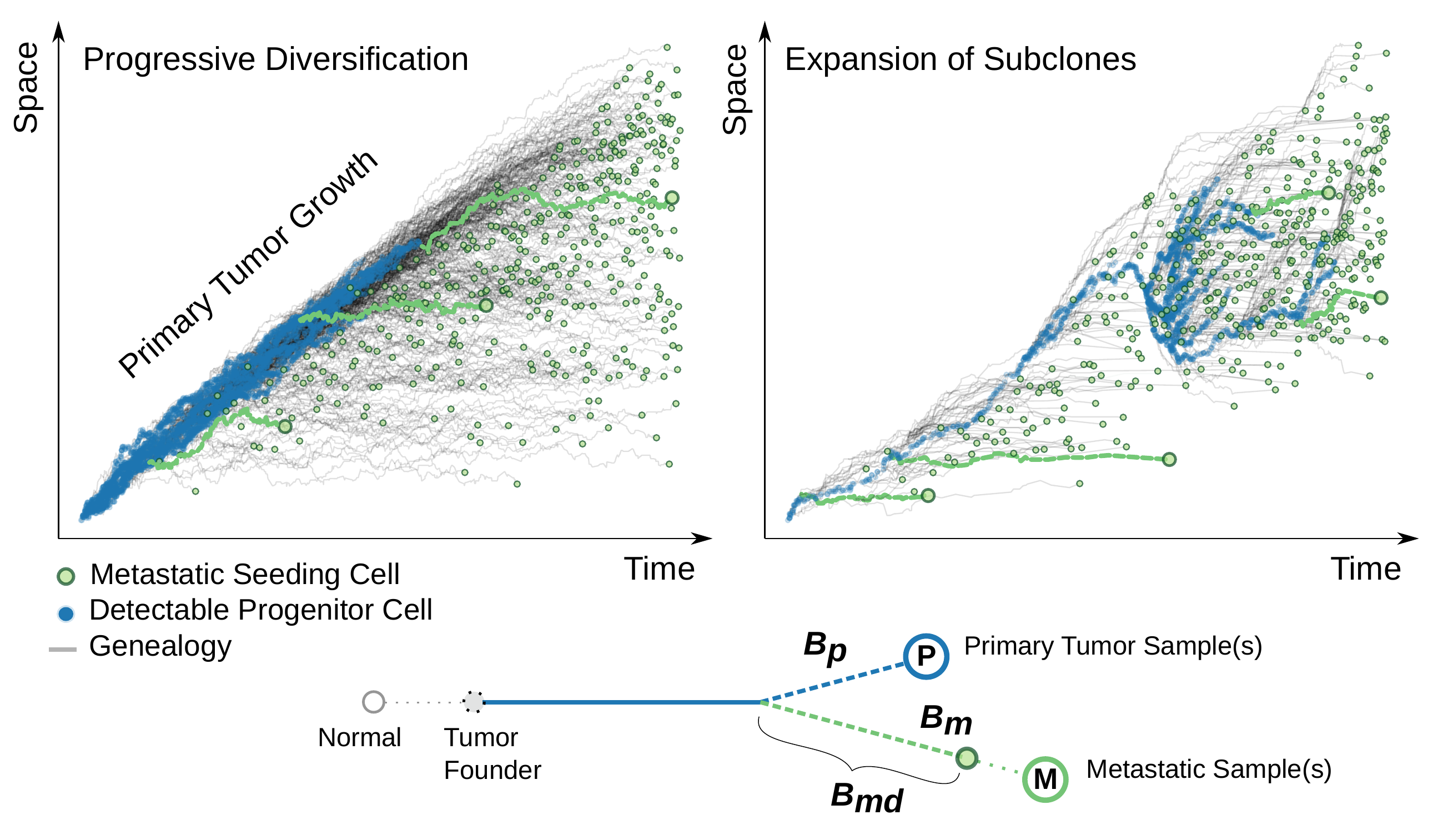
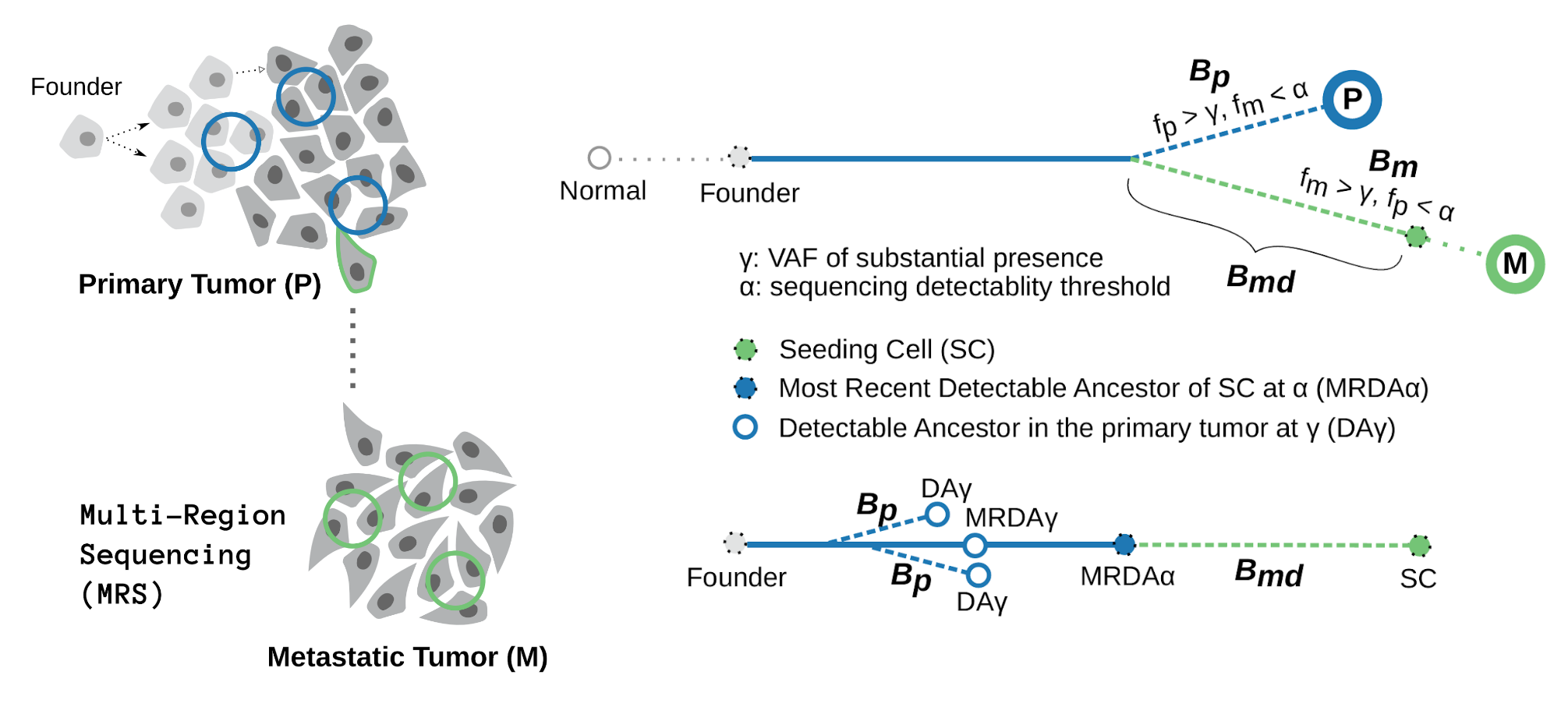
Accordingly, the subloncal SCNAs, i.e., the SCNAs that are only present in a subset of cancer cells due to ongoing chromosomal instability in tumor expansion, would have an impact on the SSNV diversification in the corresponding genomic region. The existing bulk sequencing data seems to suggest that the further SCNA evolution after tumor transformation is only detectable for a minor fraction of the genome. Although this could be due to detectablity issues and does not reflect the actual SCNA rate post transformation, we may first focus on the evolutionary history of the SCNAs dominating the cancer cell populations. In this preprint “Evolving copy number gains promote tumor expansion and bolster mutational diversification” in bioRxiv, we leveraged the quantitative bias between SCNA and SSNVs to hunt copy number gains appearing close to the onset of population expansion, and utilized a multi-type branching process model to reveal the menchanism giving rise to the late-appearing but dominating SCNAs.