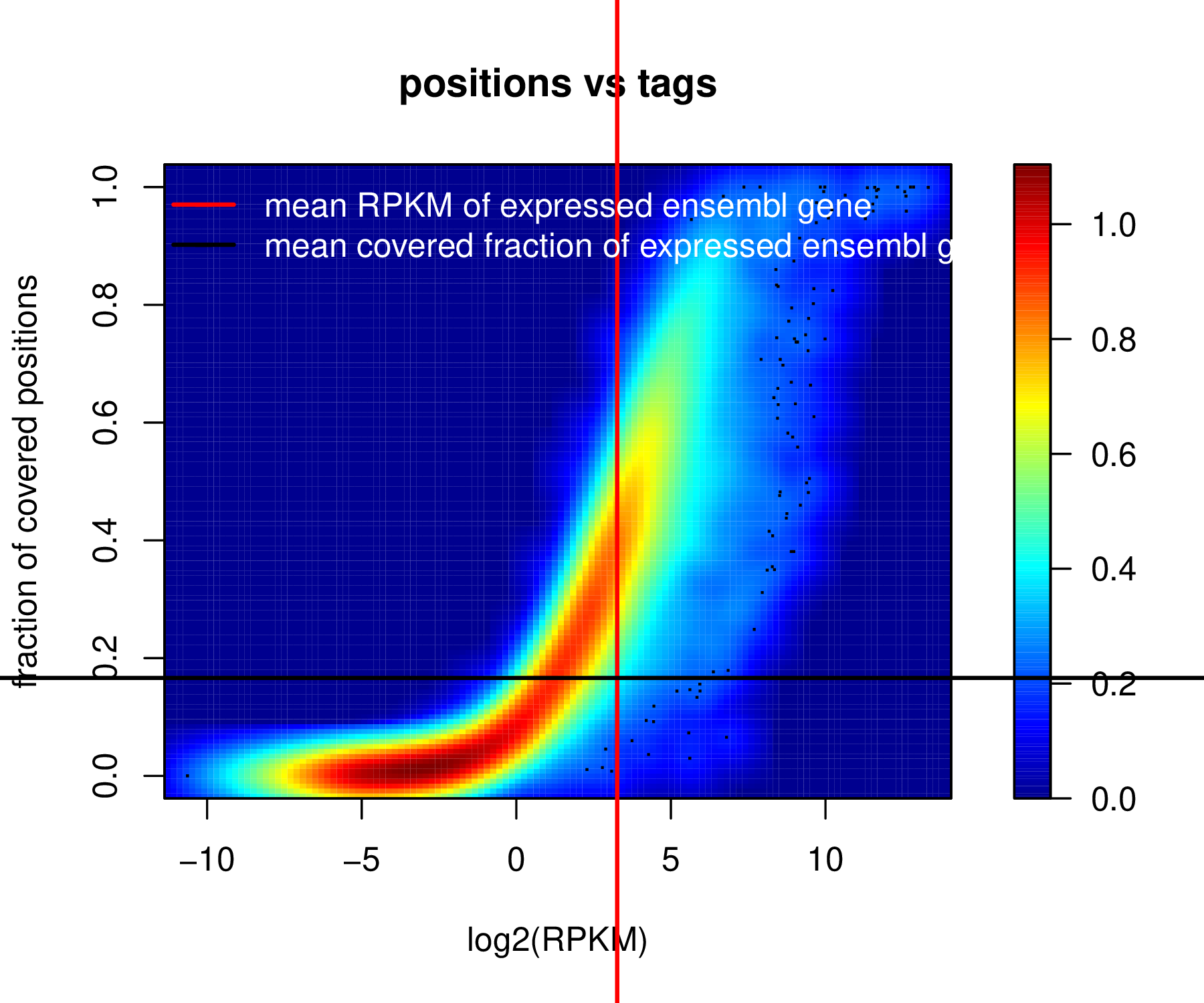
The term “coverage” for a gene in an RNA-seq experiment can be viewed in two ways. The first is the overall number of reads (depth of coverage) that are mapped to the gene, which serves to deduce a normalized “transcriptional abundance” measurement, such as the widely used R(F)PKM (Reads or Fragments Per Kilobase of transcript per Million reads mapped) and TPM (Transcripts Per Million). See a recent paper for these. However, less mentioned the other the fraction of the gene covered, more strictly, the positions in the gene where a tag starts from (fractional coverage). Plotting these two “coverage”s for each gene is an effective way to highlight an RNA-seq experiment’s quality. The rationale behind such a plot is that if the depth of coverage is high, the gene should presumably reach a high fractional coverage. If the fractional coverage remains low for very high depth of coverage, the experiment may have strong position bias which suggests a suboptimal sampling of the RNA molecules.